Menu blogs

3. Liaisons chimiques : A. Liaisons covalentes (MAO-A et rhodopsine) ->>
<<- 1. La tête dans un site actif : Le LSD couplé à son récepteur 5HT2B
2. Prédire la cardiotoxicité d'un RC ? RdR par l'IA.
19 février 2022 à 19:26
Aujourd'hui, j'aimerais vous parler d'intelligence artificielle (IA), et en profiter pour vous montrer qu'on peut potentiellement l'utiliser comme outil de réduction des risques (RdR).
L'objectif de ce billet est déjà de vous expliquer les bases de l'intelligence artificielle, puis d'appliquer cet outil pour tenter de prédire la cardiotoxicité d'une molécule n'ayant jamais été recherchée en laboratoire, par exemple un nouveau RC.
1. Le processus de développement d'un médicament
Lorsque les chercheurs essayent de concevoir un médicament, le processus passe par plusieurs phases.
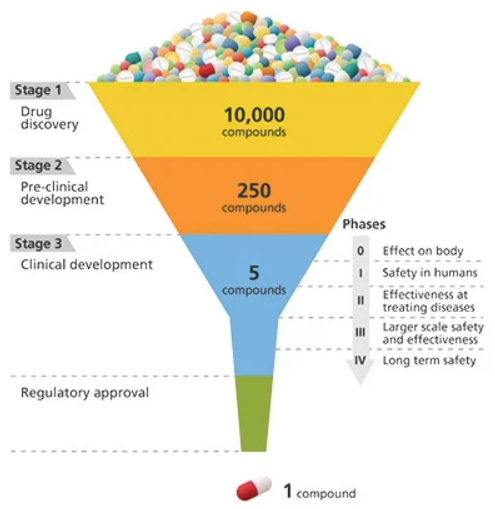
Une fois la cible identifiée, un récepteur par exemple, il est possible de tester l'interaction entre le récepteur et des milliers (voir centaines de milliers) de molécules différentes. Cela s'appelle le criblage haut débit. Les boîtes pharmaceutiques les plus fortunées ont des machines automatiques pour faire cela : elles font des dizaines de tests en éprouvette (dits tests "in vitro" avec des cellules dans des éprouvettes, à opposer à des tests "in vivo" chez des rats par exemple, ou "in silico" par simulations informatiques) avec à chaque fois le récepteur en question et une molécule différente. L'objectif est de trouver l'aiguille (la ou les molécules active sur la cible) dans une botte de foin (la banque contenant les X molécules différentes).
Si les chercheurs sont chanceux, ils vont identifier quelques centaines de molécules différentes (ils appellent ça des "hits" dans le jargon pharmaceutique) et passer à la seconde phase, le développement préclinique. Les molécules identifiées à l'étape précédentes sont optimisées, leur toxicité est testée sur des cellules, on observe leur impact sur des modèles plus complexes que des cellules, poisson zèbre, rat, souris (tests "in vivo")... Au cours de ce processus, si une molécule est très prometteuse, mais qu'elle montre des signes de toxicité, elle est immédiatement sortie du lot. Les quelques molécules qui ne sont pas éliminées sont appelées des "leads" et vont passer à l'étape suivante : la phase clinique, tests chez l'homme. Comme vous vous en doutez, seules les molécules qui ont prouvé leur non-dangerosité au maximum sont administrées chez l'homme.
2. Molécules propres, molécules sales
Une étape indispensable avant tout administration chez l'homme, c'est le test de la molécule candidate sur d'autres cibles que le récepteur d'intérêt. Certaines protéines dans le corps sont connues pour être toxiques lorsqu'elles sont activées, c'est le cas des canaux hERG (hERG pour ... "the human Ether-à-go-go-Related Gene", ne me demandez pas pourquoi). Si on se rend compte qu'une molécule active fortement les canaux hERG, même si elle est très prometteuse sur la cible en question, c'est Game Over pour elle. En effet, les molécules qui activent ces canaux peuvent créer de dangereuses arythmies cardiaques, appelées "torsades-de-pointes", qui peuvent provoquer des morts subites.
Les chercheurs préfèrent donc identifier les molécules les plus spécifiques possibles sur la cible en question. Si elles ne tapent que là où on veut qu'elles tapent, alors les risques d'effets secondaires sont grandement diminués. De là viens là le concept de molécule "sale" ou "propre". Une molécule sale, c'est une molécule qui tape sur plein de cibles différentes. L'éthanol en est une. L'ibogaïne est l'archétype de la molécule sale. Selon la concentration, elle tape sur : les récepteurs opiacés, les récepteurs sigma, les récepteurs sérotoninergiques, les récepteurs nicotiniques, les récepteurs NMDA...

Si l'on regarde rapidement sa structure, on peut voir clairement le noyau indole typique des tryptamines. Pour l'action sur les récepteurs NMDA, je ne connais pas le site actif de la mémantine et donc je peux me tromper avec mon hypothèse, mais on retrouve une partie de la mémantine dans la structure de l'ibogaïne et c'est peut être de là dont viens son côté dissociatif.
Je vous spoile direct : non contente de pouvoir activer la myriade de récepteurs dont on vient de parler, l'ibogaïne active aussi les canaux hERG, ce qui en fait une drogue potentiellement cardiotoxique.
Nous arrivons finalement à notre question initiale...
3. Comment prédire l'activité d'une molécule pour les canaux hERG ?
On peut faire ça à l'aide de l'intelligence artificielle (IA). Les chercheurs de cet article ont créé un modèle de machine learning (ML) (pour faire simple, un autre nom pour IA), et ils l'ont mis à disposition à l'aide d'un serveur web. Vous, moi, pouvons l'utiliser.
Avant d'utiliser leur webserveur, j'ai tout de même la présomption de vouloir vous expliquer très rapidement (et sans maths, juste visuellement) en quoi consiste l'IA. Ce n'est pas si compliqué, et ce n'est pas vraiment de l'intelligence (artificielle) comme vous allez le voir.
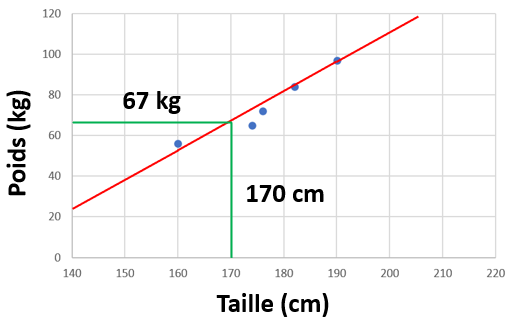
Mettons que nous avons un jeu de données sur plusieurs personnes, avec 2 informations : Leur taille en cm, leur poids en kg.
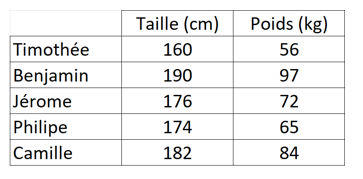
Nous pouvons mettre ces données en graphique :
Si l'on veut décrire ces données, instinctivement, on peut tout simplement tracer un trait. 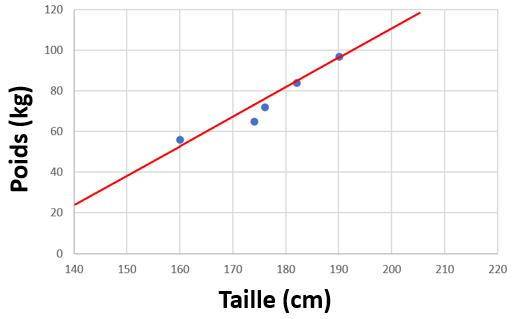
Ce trait, cette droite, c'est un modèle mathématique de notre jeu de donnée (une régression linéaire, le plus simple). Ni plus, ni moins. Selon notre jeu de données, et suivant notre modèle nouvellement créé, on peut prédire le poids qu'aurait une personne de 170 cm. Grossièrement, environ 67 kg.
Récapitulons : Grâce à un jeu de données, nous avons construit un modèle pour prédire une valeur, le poids. Ce modèle est extrêmement simple, mais tous les algorithmes de machine learning essayent de faire plus ou moins la même chose : Décrire un jeu de donnée de façon mathématique, pour pouvoir faire une prédiction sur un problème en s'appuyant sur les données existantes. 2 précisions :
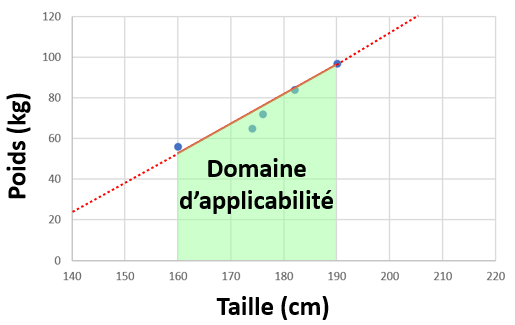
- Si nous avions eu 10000 personnes au lieu de 5 dans notre jeu de donnée, le modèle (la droite) aurait eu une meilleure performance de prédiction du poids dans la population générale. De manière générale (même si ce n'est pas toujours le cas car il y a la question de la qualité des données), plus de données, c'est des modèles plus précis.
- Si nous utilisons notre modèle linaire pour prédire le poids d'un bébé par exemple, notre modèle aura surement une performance médiocre. Nous n'avons que des personnes entre 1m60 et 1m90. Cet intervalle entre 1m60 et 1m90, c'est le domaine d'applicabilité de notre modèle. Si nous sortons de ce modèle, le modèle prédira toujours une valeur, mais la performance sera probablement médiocre.
4. Le webserveur Pred-hERG.
Il existe un énorme intérêt à réussir à prédire rapidement l'action d'une molécule sur le canal hERG. Comme dit précédement, si une molécule est fortement active sur le canal, ses chances d'être autorisé pour devenir un médicament sont faibles. Plus tôt son activité sur le canal est identifiée dans le processus de création d'un médicament, plus on évite des dépenses inutiles. Les chercheurs de l'article sus-linké ont donc créé un modèle de machine learning pour prédire cela, dans l'idée d'accélérer le processus de sélection des molécules potentielles.
Sauf qu'à la place de 5 entrées comme dans notre base de donnée taille/poids d'exemple, ils ont rassemblé les données in vitro d'environ 6000 molécules. C'est-à-dire que pour chacune de ces molécules, on sait (1) leur activité sur le canal hERG (la valeur qu'on cherche à prédire, le poids dans notre exemple), (2) on connaît la structure des 6000 molécules (la taille dans notre exemple). Le problème, c'est qu'un ordinateur ne fait pas sens d'une structure sous la forme d'une représentation visuelle. Pour contourner ce problème, il existe ce qu'on appelle des descripteurs physicochimiques, qui permettent de décrire la structure d'une molécule en un langage que l'ordinateur peut comprendre. On a par exemple la masse moléculaire de la molécule, son nombre de carbones, son nombre de doubles liaisons, de cycles... Au lieu d'un seul descripteur (la taille), ils ont généré à partir de la structure de chaque molécule des centaines de différents descripteurs.
Une fois ceci fait, ils ont "appris" (c'est-à-dire) entrainé leur modèle sur ces données, et ils l'ont mis online ici (il faut scroller en bas). Comme il aurait été pénible de devoir générer soi-même les descripteurs physico-chimiques pour chaque molécule dont on veut prédire l'affinité pour hERG, ils ont créé une interface simplifiée qui nous fait ça en coulisse. Il y a juste 2 manières de procéder : (1) Soit ajouter un SMILES d'une molécule, soit (2) la dessiner.

Un SMILES, c'est un langage symbolique qui permet de décrire (de façon extrêmement compacte, son principal intérêt) une molécule, tout en conservant un maximum d'information sur la molécule. Par exemple, la formule brute de l'ibogaine (C20H26N2O) est très compacte, mais contrairement au SMILES, elle ne contient ni l'information du nombre de doubles liaisons, ni comment la molécule s'agence dans l'espace.
On sait déjà que l'ibogaïne active le canal hERG, on va donc commencer par tester cette molécule-là pour voir quelle est la prédiction de leur modèle. Pour ce faire, sur la page wikipédia de l'ibogaïne est fourni le SMILES de la molécule.
CC[C@H]([C@@H]1[N@](C2)CC3)C[C@@H]2C[C@H]1C(N4)=C3C5=C4C=CC(OC)=C5
Voici les résultats du modèle :
On peut observer que l'ibogaïne est prédite comme potentiellement cardiotoxique, et que l'interaction entre cette molécule et les canaux hERG est prédite comme faible à modérée. Pour chaque résultat est donné un pourcentage de fiabilité de la prédiction. Très important, on peut aussi voir que la structure de l'ibogaïne est à la limite du domaine de fiabilité du modèle (si le score avait été inférieur à 0.26, on serai sorti du domaine d'applicabilité) : Pour simplifier, cela veut dire que l'ibogaïne ressemble juste assez structurellement aux 6000 molécules utilisées pour entrainer le modèle, pour que la prédiction soit fiable.
On peut tester le 3MMC, selon le modèle, le 3MMC n'as pas d'action sur le canal hERG.

Une note importante : Ce webserveur est un modèle de machine learning tout ce qu'il y a de plus expérimental. Son utilité principale (tout du moins à mon avis) réside dans sa capacité à trier rapidement des milliers de molécules dans le processus de création d'un médicament. Ce n'est aucunement parce que la prédiction du modèle d'une molécule ressort sans cardiotoxicité avérée qu'il faut considérer la molécule en question comme non dangereuse pour la consommation (vrai négatif).
J'ai été surpris que le THC soit reporté par le modèle comme potentiellement cardiotoxique, avec une forte activité sur le canal hERG.

Il est avéré que certains cannabinoïdes sont cardiotoxiques (par exemple le JWH-030 ici). La possibilité de faux positif est tout aussi possible.
En creusant un peu plus, le CBD est lui aussi reporté comme cardiotoxique par le modèle, pourtant, il aurai une action bloquante sur les canaux hERG, venant moduler la dangerosité du THC selon cet article (la nature fait bien les choses, et nous surprend chaque jours !). J'aurai appris quelque chose. Ces erreurs du modèle sont explicables par le fait que la proximité chimique entre le THC et le CBD est extrêmement forte.

En effet, la différence entre une molécule activant une protéine (agoniste), une molécule bloquante (antagoniste), ou une molécule désactivante (agoniste inverse) est souvent extrêmement subtile, et se joue parfois à un atome près (sans parler des agonistes partiels, plus sur le sujet à venir), ce qui pousse le machine learning dans ses retranchements. En l'état, leur modèle semble dont seulement pouvoir prédire si une molécule va interagir ou pas avec les cannaux hERG, pas la façon dont les molécules agissent sur le canal.
EDIT : Après coup je me rends compte que le CBD et le THC sortent du modèle d'applicabilité du modèle (encore une fois, comprendre qu'il ne doit pas y avoir de cannabinoïdes dans la base de données ayant servi à construire le modèle). La prédiction n'est donc pas fiable pour ces molécules. Cependant, la discution à propos de la difficulté de prédire la différence entre une molécule qui active ou qui bloque le canal reste entièrement valide.
C'est tout pour aujourd'hui. En espérant que ça n'ai pas été trop long ni compliqué. Je suis conscient que les concepts sont pas évidents à capter. Je serai content si vous avez un peu touché du doigt l'idée derrière l'intelligence artificielle (prédire une variable en utilisant des données pré-existantes). J'espère que vous avez compris que ce n'est ni magique, ni infaillible, et que les performances dépendent de la qualité/quantité des données, et du problème considéré. Merci à vous si vous avez réussi à lire jusqu'ici.

- Mychkine
Artichaut dans une caisse d'oranges - 27 février 2022 à 14:20
Merci beaucoup d'avoir pris le temps de rédiger ce texte tres éclairant et de grande qualité !
J'attends la suite avec beaucoup d'intéret.
Pour répondre a tes interrogations, je ne trouve pas ce billet trop long, ni trop complexe. J'aurais meme apprécié encore plus de détails sur les structures chimiques des différentes molécules testées (que signifient les points de couleurs dans les représentations en sortie de l'algo ?).
Pour l'activité NMDA de l'ibogaine, en la comparant a la kétamine :
Et au PCP :

Je me dis que le cycle benzene et le celui avec l'azote (je ne sais pas si il a un nom) de l'ibo peuvent peut-etre aussi l'expliquer.
Mychkine a écrit
Salut.
Merci beaucoup d'avoir pris le temps de rédiger ce texte tres éclairant et de grande qualité !
J'attends la suite avec beaucoup d'intéret.
Pour répondre a tes interrogations, je ne trouve pas ce billet trop long, ni trop complexe.
Super alors merci pour ton retour ! :)
J'aurais meme apprécié encore plus de détails sur les structures chimiques des différentes molécules testées (que signifient les points de couleurs dans les représentations en sortie de l'algo ?).
En fait, c'est une carte de probabilité des contributions atomiques de la molécule en question, créée par le modèle. En vert, tu as les parties de la molécule qui sont prédites comme ayant une contribution positive dans le blocage des canaux hERG. En gris, pas de contribution, et en rouge, une contribution négative en tant que bloqueur.
Grâce à une telle map, on peut tenter d'identifier la partie d'une molécule qui contribue le plus à son activité sur le canal, et donc l'optimiser pour diminuer cette activité.
Le machine learning a souvent comme problème son fonctionnement "boîte noire". On a des données en entrée, on a une prédiction en sortie, mais comment ?, pourquoi ?, ne sont pas des informations données par la plupart des modèles. Les chercheurs ont donc développé des moyens d'y voir plus clair. On a par exemple des algorithmes plus facile à comprendre, comme les arbres de décisions.
Cette fonctionnalité du modèle à fournir une carte de probabilité avec le résultat fait un peu partie de cet effort. Ça permet de mieux comprendre et illustrer la prédiction.
Le modèle présenté ici, c'est en fait une des 2 branches du machine learning : L'apprentissage supervisé. Par supervisé, on entend que le modèle est entrainé grâce à une base de données de molécules dont on connaît l'action (activité ou non sur le cannal hERG).
L'autre branche du machine learning, c'est l'apprentissage non supervisé. Dans notre cas avant de faire le modèle décris dans l'article, on aurait pu donner l'ensemble des 6000 molécules de la base de donnée à un algorithme pour tenter de voir des relations cachées dans la base de données. Simplement donner à l'algorithme la tâche de séparer les molécules en 2 groupes par exemple, sans lui dire quelle molécule est active ou non sur le canal hERG. Si le modèle arrive à bien les séparer en identifiant un groupement spécifique dans les bloqueurs hERG qui n'est pas retrouvés dans les autres molécules, alors on a généré de l'information sur le problème (ce groupement X est important dans le blocage du canal hERG). Ce que je viens de décrire s'appelle le clustering, je ne suis pas sûr de la traduction, mais c'est l'idée de laisser un algorithme regrouper les données en paquets.
Pour l'activité NMDA de l'ibogaine, en la comparant a la kétamine :
Image: https://www.psychoactif.org/forum/uploa … 1645/k.png
Et au PCP : Image: https://www.psychoactif.org/forum/uploa … 45/pcp.png
Je me dis que le cycle benzene et le celui avec l'azote (je ne sais pas si il a un nom) de l'ibo peuvent peut-etre aussi l'expliquer.
Effectivement tu as raison, on retrouve ce cycle à 6 carbones non aromatiques, avec un groupement amine. En fait ça peut faire un bon billet pour la suite, essayer de docker l'ibogaïne, la kétamine et le PCP sur les récepteurs NMDA. Je vais voir ça :)

- Morning Glory
Holofractale de l'Hypervérité - 04 mars 2022 à 12:18

- Zed41
Nouveau membre
- 05 mars 2022 à 10:11
- Zed41
Nouveau membre - 05 mars 2022 à 10:17

"Plus de contours et une intensité dans la couleur verte signifie une contribution plus importante d'un atome ou d'un fragment [de la molécule] dans l'antagonisme de hERG"
En gros plus c'est vert plus ça a de chance d'être cardiotoxique...
Tout en prenant en compte si on est ou non dans le domaine d'applicabilité...
Ainsi que la fiabilité des résultats...
Que c'est une simulation informatique (in silico je connaissais pas ça claque).
Et qu'elle ne vise que le récepteur hERG loin d'être le seul récepteur (trouvé et existant) pouvant induire une cardiotoxicité.
Bref, un super outil pour les chercheurs !
Et pourtant, c'est bien la molécule de 3MMC sur l'image de sortie du webserveur. Juste réarrangée de différente manière, mais tout y est.
Merci pour l'image d'interprétation, c'est plus clair comme cela ! :) Et oui tes précisions sont très importantes. C'est une prédiction avant tout. Leur modèles ont une assez bonne performance, mais ces approches sont présentes avant tout pour guider et orienter les tests expérimentaux in vitro / in vivo. Pas pour en tirer des conclusions fermes.
Merci de ton retour !
J'ai déjà liké un de tes posts précédemment donc je ne peux pas le faire sur celui-là, mais merci pour toutes ces explications, j'aime! Bienvenue sur PA!
Merci à toi, ça va me pousser à continuer l'expérience. À bientôt !

- Johan89
Nouveau Psycho - 11 mars 2022 à 15:13
g-rusalem a écrit
Étant débutant dans la vulgarisation, je serai intéressé d'obtenir des retours, même (surtout ?) négatifs vis-à-vis de ce billet. Trop complexe ? Trop long ? Pas assez clair ? Merci d'avance pour ceux qui pourraient m'écrire un mot. Ça me sera utile à la fois ici pour les prochains billets, et dans ma vie pro.
Pratiquant moi même beaucoup de ML dans mes études, j'ai trouvé ça une chouille frustrant de parler de régression car -même si je n'ai pas encore lu le papier d'élaboration du model- c'est certainement d'autres types d'algo de ML qui sont utilisés non? Si l'on peut estimer le risque d'interaction via une classification sous-jacentes: ("au conditionnel", j'ai envie d'utiliser du K-NN et ou SVM je dirai à première vu, où l'on ferait des découpages en classes qui représenteraient graduellement un palier de toxicité, pour ensuite essayer de recouper en exploration de données, de quelles caractéristiques disposent les molécules appartenant au même cluster). Cependant ce n'est pas un domaine que je ne connais pas bien le chemical design donc peut-être qu'en pratique cela s'applique mal).
Néanmoins je comprends que c'est pas facile de vulgariser tout ça et je pense que je serai incapable de faire mieux que toi la dessus, donc clairement la régression linéaire pour faire une analogie basique taille-masse, c'est pas mal. Juste peut-être dire en complément/précision qu'il existe une panoplie d'autres méthodes, plus pertinente pour certains sets de données spécifiques et dans certains cas. Je pense que si tu donnes les noms des méthodes/algos anecdotiquement, ceux que ça a intéressé peuvent ensuite aller faire leur propre recherches sur google pour approfondir ça.
Sinon merci de mettre ça sur psychoactif c'est cool 
Johan89 a écrit
Pratiquant moi même beaucoup de ML dans mes études, j'ai trouvé ça une chouille frustrant de parler de régression car -même si je n'ai pas encore lu le papier d'élaboration du model- c'est certainement d'autres types d'algo de ML qui sont utilisés non? Si l'on peut estimer le risque d'interaction via une classification sous-jacentes: ("au conditionnel", j'ai envie d'utiliser du K-NN et ou SVM je dirai à première vu, où l'on ferait des découpages en classes qui représenteraient graduellement un palier de toxicité, pour ensuite essayer de recouper en exploration de données, de quelles caractéristiques disposent les molécules appartenant au même cluster). Cependant ce n'est pas un domaine que je ne connais pas bien le chemical design donc peut-être qu'en pratique cela s'applique mal).
Difficile de prévoir quel modèle utiliser sur ses données. Les chercheurs testent plusieurs approches en parallèles habituellement. Y'a tout de même certains modèles qui marchent mieux si tu as peu de données. Dans ce cas, il faut privilégier des modèles assez simple sinon on risque d'overfitter ses quelques datas. Si t'as beaucoup de données, alors les approches réseaux de neurones peuvent être meilleures que les approches plus basiques. Parfois, on essaye même de coupler plusieurs modèles ensemble pour générer une prédiction, l'idée étant que l'erreur différente des modèles se compensera pour donner un modèle plus performant que les différents modèles pris séparément. Dans leur cas, ils ont utilisé un modèle QSAR (Quantitative structure activity relashionship).
Je comprends que ça t'ait laissé un peu sur ta faim ! J'aurais pu faire la même chose, mais en ajoutant des parties plus techniques pour les personnes aguerries. Merci de ton retour :)
Sinon merci de mettre ça sur psychoactif c'est cool
Merci, super content que ça t'ait tout de même plu 
- Johan89
Nouveau Psycho - 25 mars 2022 à 17:33
g-rusalem a écrit
Dans ce cas, il faut privilégier des modèles assez simple sinon on risque d'overfitter ses quelques datas.
C'est le pb classique l'overfit, en ce moment j'ai pas mal de forecasting de timeseries à faire, et des modèles trop complexes sur trop peu de données ça overfit souvent.
g-rusalem a écrit
Si t'as beaucoup de données, alors les approches réseaux de neurones peuvent être meilleures que les approches plus basiques.
Sacrément chouette le deep learning pour du chem design. Si seulement j'avais un peu plus de temps (ça viendra bientôt j'espère), pour pouvoir jeter un coup d'oeil. J'ai des tas de trucs comme ça à regarder, qui essaye de proposer des frameworks pour du deeplearning: https://pubs.acs.org/doi/10.1021/acs.jcim.0c00321
g-rusalem a écrit
Dans leur cas, ils ont utilisé un modèle QSAR (Quantitative structure activity relashionship).
Thank you pour les noms, ça me permettra de fouiller un peu : https://www.youtube-nocookie.com/embed/WjoI2ZBrV2k?t=128&feature=oembed
Question un peu indiscrète, tu bosses dans le privé ou dans le public (labo type CNRS, ou rattaché à une ENS par exemple)??? Parce que comme il y a une bonne partie de ce taff qui consiste à juste développer des frameworks ou bien des méthodes pour mieux gérer la data ça touche à quelque chose qui n'est pas directement monnayable donc ça m'intrigue.
Si c'est du privé, c'est génial de pouvoir avoir des financements pour des trucs un peu plus au niveau que de l'exploitation direct s'il s'agit de R&D dans le privé, vraiment ça donne le smile !
Question un peu indiscrète, tu bosses dans le privé ou dans le public (labo type CNRS, ou rattaché à une ENS par exemple)??? Parce que comme il y a une bonne partie de ce taff qui consiste à juste développer des frameworks ou bien des méthodes pour mieux gérer la data ça touche à quelque chose qui n'est pas directement monnayable donc ça m'intrigue.
Si c'est du privé, c'est génial de pouvoir avoir des financements pour des trucs un peu plus au niveau que de l'exploitation direct s'il s'agit de R&D dans le privé, vraiment ça donne le smile !
Hello ! Là, je suis en milieu académique, mais je ne sais pas si je vais y rester. Il y a autant d'application dans la recherche fondamentale que dans la recherche privée, que ça soit en modélisation moléculaire ou en machine learning.
- oazis
Nouveau membre - 11 juin 2022 à 11:33
g-rusalem a écrit
Aujourd'hui, j'aimerais vous parler d'intelligence artificielle (IA), et en profiter pour vous montrer qu'on peut potentiellement l'utiliser comme outil de réduction des risques (RdR).
L'objectif de ce billet est déjà de vous expliquer les bases de l'intelligence artificielle, puis d'appliquer cet outil pour tenter de prédire la cardiotoxicité d'une molécule n'ayant jamais été recherchée en laboratoire, par exemple un nouveau RC.
1. Le processus de développement d'un médicament
Lorsque les chercheurs essayent de concevoir un médicament, le processus passe par plusieurs phases.
Image: https://www.psychoactif.org/forum/uploa … 645/11.pngUne fois la cible identifiée, un récepteur par exemple, il est possible de tester l'interaction entre le récepteur et des milliers (voir centaines de milliers) de molécules différentes. Cela s'appelle le criblage haut débit. Les boîtes pharmaceutiques les plus fortunées ont des machines automatiques pour faire cela : elles font des dizaines de tests en éprouvette (dits tests "in vitro" avec des cellules dans des éprouvettes, à opposer à des tests "in vivo" chez des rats par exemple, ou "in silico" par simulations informatiques) avec à chaque fois le récepteur en question et une molécule différente. L'objectif est de trouver l'aiguille (la ou les molécules active sur la cible) dans une botte de foin (la banque contenant les X molécules différentes).
Si les chercheurs sont chanceux, ils vont identifier quelques centaines de molécules différentes (ils appellent ça des "hits" dans le jargon pharmaceutique) et passer à la seconde phase, le développement préclinique. Les molécules identifiées à l'étape précédentes sont optimisées, leur toxicité est testée sur des cellules, on observe leur impact sur des modèles plus complexes que des cellules, poisson zèbre, rat, souris (tests "in vivo")... Au cours de ce processus, si une molécule est très prometteuse, mais qu'elle montre des signes de toxicité, elle est immédiatement sortie du lot. Les quelques molécules qui ne sont pas éliminées sont appelées des "leads" et vont passer à l'étape suivante : la phase clinique, tests chez l'homme. Comme vous vous en doutez, seules les molécules qui ont prouvé leur non-dangerosité au maximum sont administrées chez l'homme.
2. Molécules propres, molécules sales
Une étape indispensable avant tout administration chez l'homme, c'est le test de la molécule candidate sur d'autres cibles que le récepteur d'intérêt. Certaines protéines dans le corps sont connues pour être toxiques lorsqu'elles sont activées, c'est le cas des canaux hERG (hERG pour ... "the human Ether-à-go-go-Related Gene", ne me demandez pas pourquoi). Si on se rend compte qu'une molécule active fortement les canaux hERG, même si elle est très prometteuse sur la cible en question, c'est Game Over pour elle. En effet, les molécules qui activent ces canaux peuvent créer de dangereuses arythmies cardiaques, appelées "torsades-de-pointes", qui peuvent provoquer des morts subites.
Les chercheurs préfèrent donc identifier les molécules les plus spécifiques possibles sur la cible en question. Si elles ne tapent que là où on veut qu'elles tapent, alors les risques d'effets secondaires sont grandement diminués. De là viens là le concept de molécule "sale" ou "propre". Une molécule sale, c'est une molécule qui tape sur plein de cibles différentes. L'éthanol en est une. L'ibogaïne est l'archétype de la molécule sale. Selon la concentration, elle tape sur : les récepteurs opiacés, les récepteurs sigma, les récepteurs sérotoninergiques, les récepteurs nicotiniques, les récepteurs NMDA...
Image: https://www.psychoactif.org/forum/uploa … 645/21.pngSi l'on regarde rapidement sa structure, on peut voir clairement le noyau indole typique des tryptamines. Pour l'action sur les récepteurs NMDA, je ne connais pas le site actif de la mémantine et donc je peux me tromper avec mon hypothèse, mais on retrouve une partie de la mémantine dans la structure de l'ibogaïne et c'est peut être de là dont viens son côté dissociatif.
Je vous spoile direct : non contente de pouvoir activer la myriade de récepteurs dont on vient de parler, l'ibogaïne active aussi les canaux hERG, ce qui en fait une drogue potentiellement cardiotoxique.
Nous arrivons finalement à notre question initiale...
3. Comment prédire l'activité d'une molécule pour les canaux hERG ?
On peut faire ça à l'aide de l'intelligence artificielle (IA). Les chercheurs de cet article ont créé un modèle de machine learning (ML) (pour faire simple, un autre nom pour IA), et ils l'ont mis à disposition à l'aide d'un serveur web. Vous, moi, pouvons l'utiliser.
Avant d'utiliser leur webserveur, j'ai tout de même la présomption de vouloir vous expliquer très rapidement (et sans maths, juste visuellement) en quoi consiste l'IA. Ce n'est pas si compliqué, et ce n'est pas vraiment de l'intelligence (artificielle) comme vous allez le voir.
Mettons que nous avons un jeu de données sur plusieurs personnes, avec 2 informations : Leur taille en cm, leur poids en kg.
Image: https://www.psychoactif.org/forum/uploa … 645/33.png
Nous pouvons mettre ces données en graphique :
Image: https://www.psychoactif.org/forum/uploa … 645/42.png
Si l'on veut décrire ces données, instinctivement, on peut tout simplement tracer un trait.
Image: https://www.psychoactif.org/forum/uploa … 645/52.pngCe trait, cette droite, c'est un modèle mathématique de notre jeu de donnée (une régression linéaire, le plus simple). Ni plus, ni moins. Selon notre jeu de données, et suivant notre modèle nouvellement créé, on peut prédire le poids qu'aurait une personne de 170 cm. Grossièrement, environ 67 kg.
Image: https://www.psychoactif.org/forum/uploa … 645/62.pngRécapitulons : Grâce à un jeu de données, nous avons construit un modèle pour prédire une valeur, le poids. Ce modèle est extrêmement simple, mais tous les algorithmes de machine learning essayent de faire plus ou moins la même chose : Décrire un jeu de donnée de façon mathématique, pour pouvoir faire une prédiction sur un problème en s'appuyant sur les données existantes. 2 précisions :
- Si nous avions eu 10000 personnes au lieu de 5 dans notre jeu de donnée, le modèle (la droite) aurait eu une meilleure performance de prédiction du poids dans la population générale. De manière générale (même si ce n'est pas toujours le cas car il y a la question de la qualité des données), plus de données, c'est des modèles plus précis.
- Si nous utilisons notre modèle linaire pour prédire le poids d'un bébé par exemple, notre modèle aura surement une performance médiocre. Nous n'avons que des personnes entre 1m60 et 1m90. Cet intervalle entre 1m60 et 1m90, c'est le domaine d'applicabilité de notre modèle. Si nous sortons de ce modèle, le modèle prédira toujours une valeur, mais la performance sera probablement médiocre.
Image: https://www.psychoactif.org/forum/uploa … 645/71.png
4. Le webserveur Pred-hERG.Il existe un énorme intérêt à réussir à prédire rapidement l'action d'une molécule sur le canal hERG. Comme dit précédement, si une molécule est fortement active sur le canal, ses chances d'être autorisé pour devenir un médicament sont faibles. Plus tôt son activité sur le canal est identifiée dans le processus de création d'un médicament, plus on évite des dépenses inutiles. Les chercheurs de l'article sus-linké ont donc créé un modèle de machine learning pour prédire cela, dans l'idée d'accélérer le processus de sélection des molécules potentielles.
Sauf qu'à la place de 5 entrées comme dans notre base de donnée taille/poids d'exemple, ils ont rassemblé les données in vitro d'environ 6000 molécules. C'est-à-dire que pour chacune de ces molécules, on sait (1) leur activité sur le canal hERG (la valeur qu'on cherche à prédire, le poids dans notre exemple), (2) on connaît la structure des 6000 molécules (la taille dans notre exemple). Le problème, c'est qu'un ordinateur ne fait pas sens d'une structure sous la forme d'une représentation visuelle. Pour contourner ce problème, il existe ce qu'on appelle des descripteurs physicochimiques, qui permettent de décrire la structure d'une molécule en un langage que l'ordinateur peut comprendre. On a par exemple la masse moléculaire de la molécule, son nombre de carbones, son nombre de doubles liaisons, de cycles... Au lieu d'un seul descripteur (la taille), ils ont généré à partir de la structure de chaque molécule des centaines de différents descripteurs.
Une fois ceci fait, ils ont "appris" (c'est-à-dire) entrainé leur modèle sur ces données, et ils l'ont mis online ici (il faut scroller en bas). Comme il aurait été pénible de devoir générer soi-même les descripteurs physico-chimiques pour chaque molécule dont on veut prédire l'affinité pour hERG, ils ont créé une interface simplifiée qui nous fait ça en coulisse. Il y a juste 2 manières de procéder : (1) Soit ajouter un SMILES d'une molécule, soit (2) la dessiner.
Image: https://www.psychoactif.org/forum/uploa … 1645/8.pngUn SMILES, c'est un langage symbolique qui permet de décrire (de façon extrêmement compacte, son principal intérêt) une molécule, tout en conservant un maximum d'information sur la molécule. Par exemple, la formule brute de l'ibogaine (C20H26N2O) est très compacte, mais contrairement au SMILES, elle ne contient ni l'information du nombre de doubles liaisons, ni comment la molécule s'agence dans l'espace.
On sait déjà que l'ibogaïne active le canal hERG, on va donc commencer par tester cette molécule-là pour voir quelle est la prédiction de leur modèle. Pour ce faire, sur la page wikipédia de l'ibogaïne est fourni le SMILES de la molécule.
CC[C@H]([C@@H]1[N@](C2)CC3)C[C@@H]2C[C@H]1C(N4)=C3C5=C4C=CC(OC)=C5
Voici les résultats du modèle :
Image: https://www.psychoactif.org/forum/uploa … 645/91.pngOn peut observer que l'ibogaïne est prédite comme potentiellement cardiotoxique, et que l'interaction entre cette molécule et les canaux hERG est prédite comme faible à modérée. Pour chaque résultat est donné un pourcentage de fiabilité de la prédiction. Très important, on peut aussi voir que la structure de l'ibogaïne est à la limite du domaine de fiabilité du modèle (si le score avait été inférieur à 0.26, on serai sorti du domaine d'applicabilité) : Pour simplifier, cela veut dire que l'ibogaïne ressemble juste assez structurellement aux 6000 molécules utilisées pour entrainer le modèle, pour que la prédiction soit fiable.
On peut tester le 3MMC, selon le modèle, le 3MMC n'as pas d'action sur le canal hERG.
Image: https://www.psychoactif.org/forum/uploa … 645/10.pngUne note importante : Ce webserveur est un modèle de machine learning tout ce qu'il y a de plus expérimental. Son utilité principale (tout du moins à mon avis) réside dans sa capacité à trier rapidement des milliers de molécules dans le processus de création d'un médicament. Ce n'est aucunement parce que la prédiction du modèle d'une molécule ressort sans cardiotoxicité avérée qu'il faut considérer la molécule en question comme non dangereuse pour la consommation (vrai négatif).
J'ai été surpris que le THC soit reporté par le modèle comme potentiellement cardiotoxique, avec une forte activité sur le canal hERG.
Image: https://www.psychoactif.org/forum/uploa … 45/111.pngIl est avéré que certains cannabinoïdes sont cardiotoxiques (par exemple le JWH-030 ici). La possibilité de faux positif est tout aussi possible.
En creusant un peu plus, le CBD est lui aussi reporté comme cardiotoxique par le modèle, pourtant, il aurai une action bloquante sur les canaux hERG, venant moduler la dangerosité du THC selon cet article (la nature fait bien les choses, et nous surprend chaque jours !). J'aurai appris quelque chose. Ces erreurs du modèle sont explicables par le fait que la proximité chimique entre le THC et le CBD est extrêmement forte.
Image: https://www.psychoactif.org/forum/uploa … 645/12.pngEn effet, la différence entre une molécule activant une protéine (agoniste), une molécule bloquante (antagoniste), ou une molécule désactivante (agoniste inverse) est souvent extrêmement subtile, et se joue parfois à un atome près (sans parler des agonistes partiels, plus sur le sujet à venir), ce qui pousse le machine learning dans ses retranchements. En l'état, leur modèle semble dont seulement pouvoir prédire si une molécule va interagir ou pas avec les cannaux hERG, pas la façon dont les molécules agissent sur le canal.
EDIT : Après coup je me rends compte que le CBD et le THC sortent du modèle d'applicabilité du modèle (encore une fois, comprendre qu'il ne doit pas y avoir de cannabinoïdes dans la base de données ayant servi à construire le modèle). La prédiction n'est donc pas fiable pour ces molécules. Cependant, la discution à propos de la difficulté de prédire la différence entre une molécule qui active ou qui bloque le canal reste entièrement valide.
C'est tout pour aujourd'hui. En espérant que ça n'ai pas été trop long ni compliqué. Je suis conscient que les concepts sont pas évidents à capter. Je serai content si vous avez un peu touché du doigt l'idée derrière l'intelligence artificielle (prédire une variable en utilisant des données pré-existantes). J'espère que vous avez compris que ce n'est ni magique, ni infaillible, et que les performances dépendent de la qualité/quantité des données, et du problème considéré. Merci à vous si vous avez réussi à lire jusqu'ici.
Le fait que le THC soit reporté par ce modèle comme une molécule cardio toxique corrèle t'il avec les observations cliniques faites à ce propos ? voici, par exemple, un article à ce sujet https://www.edimark.fr/Front/frontpost/ … /13535.pdf
Effectivement le THC est bel et bien potentiellement cardiotoxique (tous comme certains cannabinoïdes synthétiques), il existe des articles là-dessus ce que je ne savais pas au moment d'écrire l'article. Ce qui m'a encore plus surpris, c'est d'apprendre que le CBD pouvait contrer l'effet néfaste du THC à ce niveau. C'est là qu'on se rend compte de la complexité de la nature.
Cependant, comme le THC et le CBD sortent du domaine d'applicabilité du modèle présenté ici, ils n'ont pas dû mettre ces données sur le THC dans leur modèle. L'étape la plus longue et la plus importante dans le machine learning (et la plus chiante, avis perso), c'est de rassembler les données, et les sélectionner par différents processus. Peut-être qu'ils ont choisi de ne pas inclure les données expérimentales du THC dans leur modèle, car ils ont jugé la qualité des données trop faibles. Ou alors ils ne sont pas tombés dessus ou n'étaient pas intéressés par la base structurelle (le scaffold) des molécules telles que le THC, le CBD et les cannabinoïdes. Toutes ces explications sont plausibles.
Merci pour ton commentaire ! (par contre de manière générale tu n'es pas obligé de quote l'entièreté du post, tu peux supprimer la majorité et ne garder que le niveau sur lequel tu réagis).

- Mister No
Pussy time - 25 août 2022 à 17:26
Une note importante : Ce webserveur est un modèle de machine learning tout ce qu'il y a de plus expérimental. Son utilité principale (tout du moins à mon avis) réside dans sa capacité à trier rapidement des milliers de molécules dans le processus de création d'un médicament. Ce n'est aucunement parce que la prédiction du modèle d'une molécule ressort sans cardiotoxicité avérée qu'il faut considérer la molécule en question comme non dangereuse pour la consommation (vrai négatif).
Du coup, on s'aperçoit que le contraire peut être vrai, avec des faux positifs.
C'est important de prendre des pincettes avec ce type d'IA. Deep learning.
Ce type d'IA a tué lors des premiers variants COVID.
Quand l'IHU Marseille à communiqué sur la prise en charge par oxygénation sans intubation, pendant très longtemps des hôpitaux ont continué à intuber les malades au prétexte que leur saturation ne s'améliorait pas assez vite.
La nouveauté de la maladie, des symptômes pulmonaires a berné l'IA et ceux qui l'ont suivi car c'est presque parfois devenu un standard.
Éviter l'incubation a évité bon nombre de décès et de complications en réa.
(Pour ceux qui se souviennent des masques décathlon qui ont sauvé des vies)
Il paraît que l'IA de Google a engagé un avocat pour ne pas riquer d'être débranché.
Mister No a écrit
Vu les circonstances, je suis venu furter.
La personne a été bannie, il me semble (pour quelles raisons ?) donc n'espérez pas de réponse. Mais je peux humblement tenter une réponse, Mr. No, si vous me le permettez.
C'est important de prendre des pincettes avec ce type d'IA. Deep learning.
Ce type d'IA a tué lors des premiers variants COVID.
Quand l'IHU Marseille à communiqué sur la prise en charge par oxygénation sans intubation, pendant très longtemps des hôpitaux ont continué à intuber les malades au prétexte que leur saturation ne s'améliorait pas assez vite.
La nouveauté de la maladie, des symptômes pulmonaires a berné l'IA et ceux qui l'ont suivi car c'est presque parfois devenu un standard.
Éviter l'incubation a évité bon nombre de décès et de complications en réa.
(Pour ceux qui se souviennent des masques décathlon qui ont sauvé des vies)
Il paraît que l'IA de Google a engagé un avocat pour ne pas riquer d'être débranché.
Il est effectivement important de comprendre les limites de son modèle. Vous décrivez le problème de l'évolution des données au cours du temps. Un cas sur lequel le modèle n'as pas été entrainé, qui sort donc du domaine d'applicabilité du modèle, et c'est une prédiction avec une faible qualité qui risque de sortir. Il existe des façons de procéder pour "updater" en temps réel le modèle. C'est ce genre de procédé qui est utilisé pour la détection du spam. Vous aidez le modèle à chaque fois que vous identifiez un mail comme du spam, car le modèle prend en compte cette nouvelle information de façon dynamique.
Ce problème que vous amenez met une autre limitation du machine learning sur la table. Il suffit de montrer une fois à un enfant qu'un arbre ça s'appelle "arbre", pour qu'il reconnaisse instantanément et pour le restant de sa vie un arbre comme un "arbre" (qu'il puisse correctement classifier un percept visuel en tant qu'arbre). Les modèles de machine learning actuels ne savent encore pas faire ça à l'heure actuelle, ou mal. Il faut beaucoup de cas à chaque nouveauté rencontrée. Dans le cas de la COVID en tout début de crise, même si l'on s'était rendu compte du nouveau problème, l'absence d'une grande quantité de données aurait rendu difficile l'update du modèle en question.
Je finis par dire qu'il y a une dernière limitation que je vois dans votre post. Vous vous arrêtez à "l'IA a tué". Combien de vies ont été sauvées grâce à l'adoption de cette technologie par les hôpitaux ? Je vous donne un autre exemple : Dès que l'IA tuera potentiellement moins de personne sur les routes que les humains, alors il sera éthiquement douteux de laisser un humain conduire. C'est juste une question de statistique et de pragmatisme.
Si l'on regarde le monde des échecs et de l'IA, les performances les plus importantes sont obtenues lorsqu'on mélange à la fois les prédictions des meilleurs joueurs d'échec humain et l'IA, Vs IA uniquement ou meilleurs joueurs humains uniquement. C'est un phénomène que l'on peut comprendre lorsqu'on utilise des "multimodèles" pour prédire une valeur. Il est parfois plus efficace de combiner plusieurs algorithmes de prédictions, pour que les erreurs de chaque modèle se compensent et améliorent le score du métamodèle (ceci est particulièrement prévalent sur les petits jeux de données ou le problème de l'overfitting doit être compensé). 1+1 = 3. Merci Bernard Weber.
- Mister No
Pussy time - 29 août 2022 à 15:25
Je finis par dire qu'il y a une dernière limitation que je vois dans votre post. Vous vous arrêtez à "l'IA a tué". Combien de vies ont été sauvées grâce à l'adoption de cette technologie par les hôpitaux ? Je vous donne un autre exemple : Dès que l'IA tuera potentiellement moins de personne sur les routes que les humains, alors il sera éthiquement douteux de laisser un humain conduire. C'est juste une question de statistique et de pragmatisme.
Sauf que l'IA s'est plantée sur ce coup.
Je ne dis pas que l 'IA de type deep learning n'est pas utile, je mettais juste en exergue les dérives quand les médecins s'effacent au profit de l'interprétation possible de l'évolution de la sat en oxygene.
 Soutenez PsychoACTIF
Soutenez PsychoACTIFAffichage Bureau - A propos de Psychoactif - Politique de confidentialité - CGU - Contact -